La TETRALOGIE DE FALLOT
Description
C'est Arthur Fallot qui le premier, en 1888, décrivit la tétralogie, rassemblant :
- une communication interventriculaire (CIV)
- une aorte à cheval sur le ventricule droit et gauche
- une hypertrophie (épaississement) du ventricule droit
- un obstacle au niveau de la voie d'éjection du ventricule droit (VD), obstacle pouvant se situer au niveau
de la valve pulmonaire (sténose valvulaire), en dessous ou au-dessus de la valve pulmonaire (sténose sous -ou supra-
valvulaire pulmonaire) ou au niveau des artères pulmonaires (PA) (ou partout à la fois).
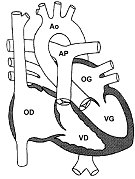 figure 1 |
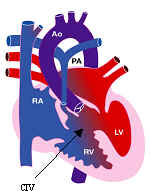 figure 2 |
figure 2 : schéma de la tétralogie de Fallot. On visualise la communication interventriculaire (CIV) entre les ventricules. L'aorte est à
cheval sur les 2 ventricules ce qui permet le mélange de sang bleu et rouge au niveau de l'aorte.
Il peut exister d'autres malformations associées comme :
- une communication interauriculaire (CIA, communication entre l'oreillette droite (RA) et l'oreillette gauche (LA))
- des communications interventriculaires additionnelles
- un arc aortique droit
- des anomalies des artères coronaires
- un canal artériel encore perméable (petit canal reliant l'aorte et les artères pulmonaires, indispensable pendant la
vie foetale)
Elle représente à peu près 10 % des malformations cardiaques congénitales.
Le plus souvent la tétralogie de Fallot est isolée,
mais parfois elle peut s'intégrer dans des syndromes (association de plusieurs signes cliniques) :
- syndrome de Down ou trisomie 21
- syndrome de la microdélétion 22
- syndrome de Goldenhar.
Présentation clinique
La disposition de l'aorte, le rétrécissement de la voie pulmonaire et la communication interventriculaire favorisent le passage de
sang bleu (non oxygéné) vers l'aorte alors que normalement l'aorte ne doit recevoir que du sang rouge (oxygéné). La présence de sang
rouge et de sang bleu au niveau de l'aorte donne un aspect "bleu" ou "cyanosé" au patient.
Cette "cyanose" sera variable d'un patient à l'autre : en effet, le degré d'obstacle au niveau de la voie pulmonaire sera variable
d'un patient à l'autre. S'il est peu important, le sang bleu se mélangera peu au sang rouge et le patient sera très peu, voire pas du tout,
cyanosé. Lorsque l'obstacle est très sévère, la cyanose pourra être très profonde. Dans certains cas, il n'y a même pas du tout d'ouverture
de la valve pulmonaire. On parle alors d'atrésie de la valve pulmonaire. Le canal artériel est alors indispensable pour permettre le passage
de sang vers les poumons.
Chez un même patient la cyanose peut également varier d'un moment à l'autre. En général, la cyanose a tendance à s'accroître avec la croissance
de l'enfant car la valve pulmonaire et les artères pulmonaires grandissent moins que le reste. Par ailleurs, l'obstacle en dessous de la valve
(sténose sous-valvulaire pulmonaire), lorsqu'il existe, est essentiellement formé par du muscle. Lorsqu'un patient s'énerve, pleure ou accélère
son coeur pour d'autres raisons, le muscle a tendance à s'épaissir et à empêcher le passage du sang vers le poumon. Le patient peut alors devenir
très bleu et même perdre connaissance par défaut d'oxygénation du cerveau. C'est ce qu'on appelle la "crise de Fallot" ou crise "hypoxique" (hypoxie
= manque d'oxygène). Cette crise peut être arrêtée en appuyant avec les jambes pliées du bébé sur son ventre. Ceci permet par plusieurs mécanismes de
favoriser le passage du sang vers les poumons et donc de rendre l'enfant plus rose. Les grands enfants (non opérés) adoptent par ailleurs
spontanément la position du "squatting" (accroupissement) lorsqu'ils font une crise hypoxique, pour les mêmes raisons. Les crises hypoxiques peuvent
également être favorisées par un bain chaud, de la fièvre, etc...
La prise en charge médicale
Le diagnostic de la tétralogie de Fallot peut se faire pendant la grossesse (dès 18-20 semaines de grossesse). Dans ces cas, il est souvent préférable
que l'enfant naisse dans un centre qui dispose d'une équipe de cardiologie pédiatrique.
Le diagnostic est cependant encore généralement fait après la naissance. A l'examen clinique les médecins pourront ausculter un souffle cardiaque et constater
la "cyanose". Celle-ci pourra être vérifiée par la mesure de la "saturation percutanée" au moyen d'un petit capteur lumineux, tout à fait indolore. Normalement,
celui-ci doit indiquer un chiffre au-delà de 95 % (correspondant à un pourcentage supérieur de 95 % d'hémoglobine (molécule transportant l'oxygène dans les globules rouges)
saturée en oxygène). En cas de cyanose, la saturation descend en dessous de 95 %. L'administration d'oxygène ne change rien à ce chiffre, et n'est donc en général pas
nécessaire. Le diagnostic sera fait par l'échocardiographie qui, en 2003, permet un diagnostic très complet de la cardiopathie. Un électrocardiogramme et une
radiographie du thorax font en général partie du bilan initial car ceux-ci donnent d'autres informations complémentaires. Si l'échocardiographie ne donne pas toutes
les informations nécessaires au chirurgien, le bilan sera complété ultérieurement par un cathétérisme cardiaque. Celui-ci consiste à, sous anesthésie générale, monter
dans les différentes cavités du coeur au moyen de "sondes ou cathéters", afin de mesurer les pressions et l'oxygénation du sang et d'injecter du produit de contraste
permettant de visualiser les différentes parties du coeur. Si d'autres anomalies non-cardiaques sont présentes, un bilan génétique à la recherche d'anomalies
chromosomiques ou génétiques sera réalisé.
La correction de la tétralogie de Fallot requiert une intervention chirurgicale. En 2003, nous proposons la cure chirurgicale vers l'âge de 6 à 9 mois. En effet, avant cela,
la chirurgie est plus difficile et donc plus risquée. Le nouveau-né est plus fragile et il est plus difficile pour le chirurgien d'opérer des tous petits enfants. La correction
implique la fermeture de la communication interventriculaire par un patch (le plus souvent en matériel synthétique) et l'ouverture de la voie pulmonaire (résection de la
sténose sous-valvulaire et supra-valvulaire pulmonaire, ouverture de la valve pulmonaire). Si la valve pulmonaire est vraiment très petite, celle-ci doit parfois être remplacée
par une valve de donneur (homogreffe) ou d'animal (hétérogreffe). Cette intervention est une chirurgie à coeur ouvert qui nécessite la mise en place d'une circulation
extracorporelle (machine coeur-poumons) pour permettre la circulation et l'oxygénation du sang pendant la chirurgie. La mortalité est à l'heure actuelle devenue faible (inférieur à 5 %).
L'intervention est obligatoirement suivie d'un passage aux soins intensifs de plusieurs jours.
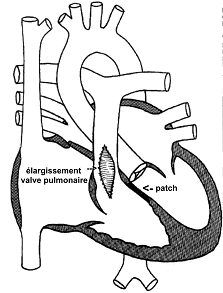
figure 3
figure 3 montrant la correction chirurgicale : fermeture de la CIV par patch et élargissement de la valve pulmonaire le plus souvent également par
interposition d'un patch.
Cependant, chez certains patients, il n'est pas prudent de réaliser la chirurgie de correction d'emblée. En effet, certains enfants naissent avec un degré de cyanose
important nécessitant une prise en charge dans les premiers mois. D'autres ont des artères pulmonaires trop petites pour permettre une correction. Dans ces 2 cas, une
intervention palliative sera d'abord réalisée. Celle-ci consiste à interposer entre l'aorte et l'artère pulmonaire un petit tuyau (shunt de Blalock) permettant d'augmenter la
perfusion des poumons et de diminuer la cyanose. Celle-ci permettra à l'enfant et aux artères pulmonaires de grandir et permettra de proposer la correction dans de meilleures
conditions.
Certains enfants sont traités par médicaments avant l'intervention. En effet, lorsque la sténose sous-valvulaire pulmonaire est importante, on sait qu'elle peut prédisposer
aux "crises de Fallot" comme expliqué ci-dessus. Dans ces conditions, un traitement par "beta-bloquants" (propanolol - Inderal®) peut être prescrit. En effet, ces "beta-bloquants"
ralentissent le coeur et diminuent la contractilité du coeur réduisant par là le risque d'épaississement du muscle provoquant la sténose sous-valvulaire. Cependant, il est important de
savoir que lorsque l'enfant fait une crise de Fallot, il est indispensable de contacter immédiatement un cardiologue pédiatre et même préférable de se rendre rapidement vers un
centre médical. Les parents peuvent aider l'enfant en le calmant le mieux possible et en réalisant le geste décrit ci-dessus (plier les jambes sur le ventre). Cependant, il sera dans
certains cas nécessaire d'administrer des médicaments par voie intra-veineuse afin de calmer l'enfant et d'améliorer le degré d'oxygénation. La survenue d'une vraie crise hypoxique
amènera en général les médecins à proposer un traitement chirurgical de façon plus ou moins urgente.
Quel avenir pour les patients opérés de tétralogie de Fallot ?
La plupart des patients opérés de tétralogie de Fallot ont une qualité de vie pour ainsi dire normale. L'espérance de vie est très bonne. En effet, dans une étude récente réalisée dans
notre centre (Bruxelles), étude revoyant 191 patients opérés entre 1964 et 1984, la survie était de 90 %, 30 ans après l'intervention (souvent réalisée plus tardivement à cette époque).
Les nombreux progrès réalisés dans les différents aspects de la prise en charge de ces patients nous permettent d'espérer des chiffres encore meilleurs pour les patients pris en charge plus
récemment. Un suivi cardiovasculaire régulier reste cependant indispensable dans tous les cas.
La pratique du sport est autorisée chez la majorité des patients. A noter toutefois, que de façon générale, nos préconisons aux patients opérés à coeur ouvert d'éviter de pratiquer du sport
à un niveau de très haute compétition. Une activité sportive régulière, même dans des clubs ayant acquis un bon niveau, reste la plupart du temps tout à fait possible.
Pour les femmes, des grossesses peuvent être envisagées chez la plupart des patientes. Un suivi cadiologique régulier par un médecin spécialisé dans les cardiopathies congénitales devra
être assuré pendant toute la grossesse et dans les mois qui suivent. En effet, la grossesse représente une situation de "stress" important pour le système cardiovasculaire chez toute
femme et ceci justifie le suivi régulier chez quelqu'un ayant bénéficié antérieurement d'une chirurgie cardiaque.
Une nuance est toutefois à apporter pour les patients ayant dès le départ des artères pulmonaires très petites ("hypoplasiques"). Même après chirurgie correctrice, si celle-ci est possible, le
ventricule droit devra assumer un travail plus important que normalement, en raison du petit diamètre des artères, ce qui peut rendre ces patients peu tolérants à l'effort.
Dans les 20 ans qui suivent l'opération initiale, 10-15 % des patients nécessiteront une nouvelle intervention. En effet, le chirurgien, lors de la première intervention, doit fréquemment élargir la
valve pulmonaire, trop étroite au départ. Cet élargissement engendre une fuite de la valve qui normalement doit être compétente. Cette fuite de la valve peut, à la longue, dilater et fatiguer le
ventricule droit, ce qui causera une fatigue à l'effort et parfois des troubles du rythme (tachycardies) qui peuvent être graves. En cas de mauvaise tolérance de cette fuite, les médecins
proposeront un remplacement de la valve pulmonaire par homogreffe ou hétérogreffe (voir ci-dessus) afin de rectifier cette situation.
En conclusion, que peut-on retenir ?
La tétralogie de Fallot est une malformation du coeur qui, à l'heure actuelle, est bien connue par le corps médical. Le diagnostic et la prise en charge peuvent se faire de façon très précoce et
permettent, dans la plupart des cas, d'envisager un avenir proche de la normale. Certains patients nécessiteront une réintervention plus tardive et ceci justifie pour tous les patients un suivi
régulier par un médecin spécialisé dans le domaine des cardiopathies congénitales. Dans des formes plus extrêmes, par exemple avec vaisseaux pulmonaires très petits ou en cas de malformations
non-cardiaques associées, le pronostic peut rester plus réservé.
(dossier réalisé par le Professeur Caroline Ovaert)